Data process
METdb data process relied on two independent workflows described using the Common workflow language (CWL) management system (https://www.commonwl.org) available here. The transcriptome assembly workflow has been used to generate assembled transcriptomes available in METdb. The assembled transcriptomes were then submitted to the EBI annotation pipeline to process the annotation step and provide the links to the annotation descriptions.
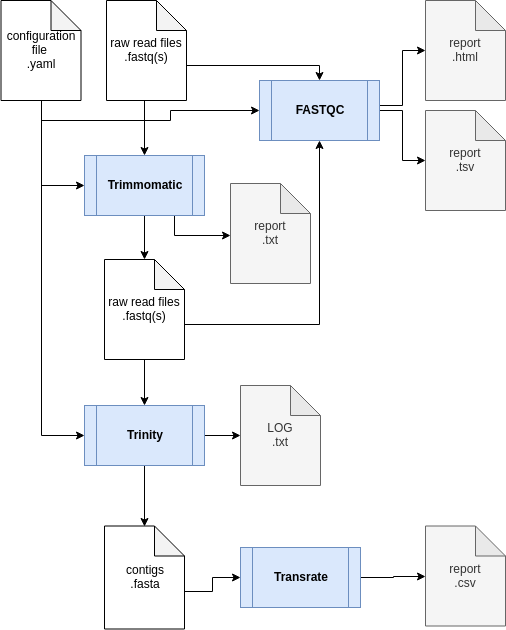
The transcriptome assembly workflow:
It includes 5 distinct steps:- Quality evaluation of raw data with FastQC;
- Raw data processing with Trimmomatic to filter and trim reads according to their sequence quality;
- Readset comparison using Simka to detect possible cross libraries contaminations;
- De novo assembly step using Trinity ;
- Quality evaluation of the assembled transcripts using Transrate .
Github repository:
- Assembly pipeline for paired-end reads: TranscriptomeAssembly-wf.paired-end.cwl
- Assembly pipeline for single-end reads: TranscriptomeAssembly-wf.single-end.cwl
CWL viewer:
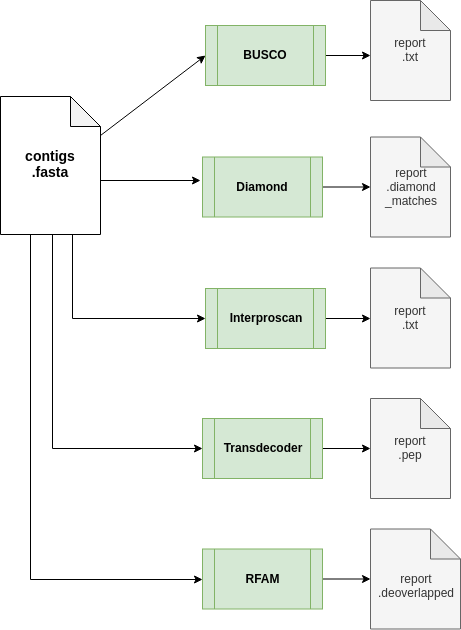
The annotation workflow:
Downstream analyses of assemblies includes:- Transcriptome completion evaluation using Busco;
- Diamond a sequence aligner for protein and translated DNA searches
- Prediction of coding regions prediction using TransDecoder;
- cmsearch uses the covariance model (CM) in cmfile to search for homologous RNAs in seqfile, and outputs high-scoring alignments
- Functional annotation of predicted proteins using the InterProscan pipeline from EMBL-EBI.
Github repository:
- Annotation pipeline for both single and paired-end reads: TranscriptsAnnotation-i5only-wf.cwl
CWL viewer: